Overview
Here we present GenomeFLTR (Dotan, et al., 2023), a web server that easily filters genomic reads. No technical skill, downloading, or computational power is needed. Raw reads are uploaded to the server and contaminated reads are removed, based on similarity to databases that are periodically and automatically updated. A user can also provide a tailored dataset to compare against. The contaminated reads are analyzed, e.g., the reads taxonomy distribution is provided. Our server provides a simple and interactive graphical user interface (GUI) that allows controlling the filtering process.
Input
The minimal input to the GenomeFLTR server consists of:
1. A single read file or two pair-ended read files. Standard formats such as Fastq and Fasta are accepted, or compressed gz versions of these two formats. The number of reads is limited to 50 millions for paired reads and 100 millions for single reads.
2. A database against which the reads are queried (e.g., to detect bacterial contaminants, a user can choose a bacterial database containing multiple genomes from a diverse set of bacteria). A user may also input a custom database. The entire set of sequence databases available in GenomeFLTR is automatically updated monthly from NCBI. These databases are processed for the Kraken search engine format (Wood, et al., 2019). Default databases are bacteria, human, fungi, protozoa, univec (i.e., a dataset of vector sequences), plasmid, archaea, viral, Kraken standard (i.e., all complete bacterial, archeal, and viral genomes in Refseq), and custom. For the custom database, a user inserts the NCBI taxonomy identifiers of the species included in Refseq (NCBI Reference Sequence Database) to compare against and may choose specific accession numbers of genomes from this species to analyze. If accession numbers are not provided, the first three genomes from Refseq are downloaded for each species.
Methodology
Three simple steps to use GenomeFLTR: (1) Upload read data and specify optional parameters; (2) View and analyze contamination results with the interactive GUI; (3) Download the filtered reads (figure 1).
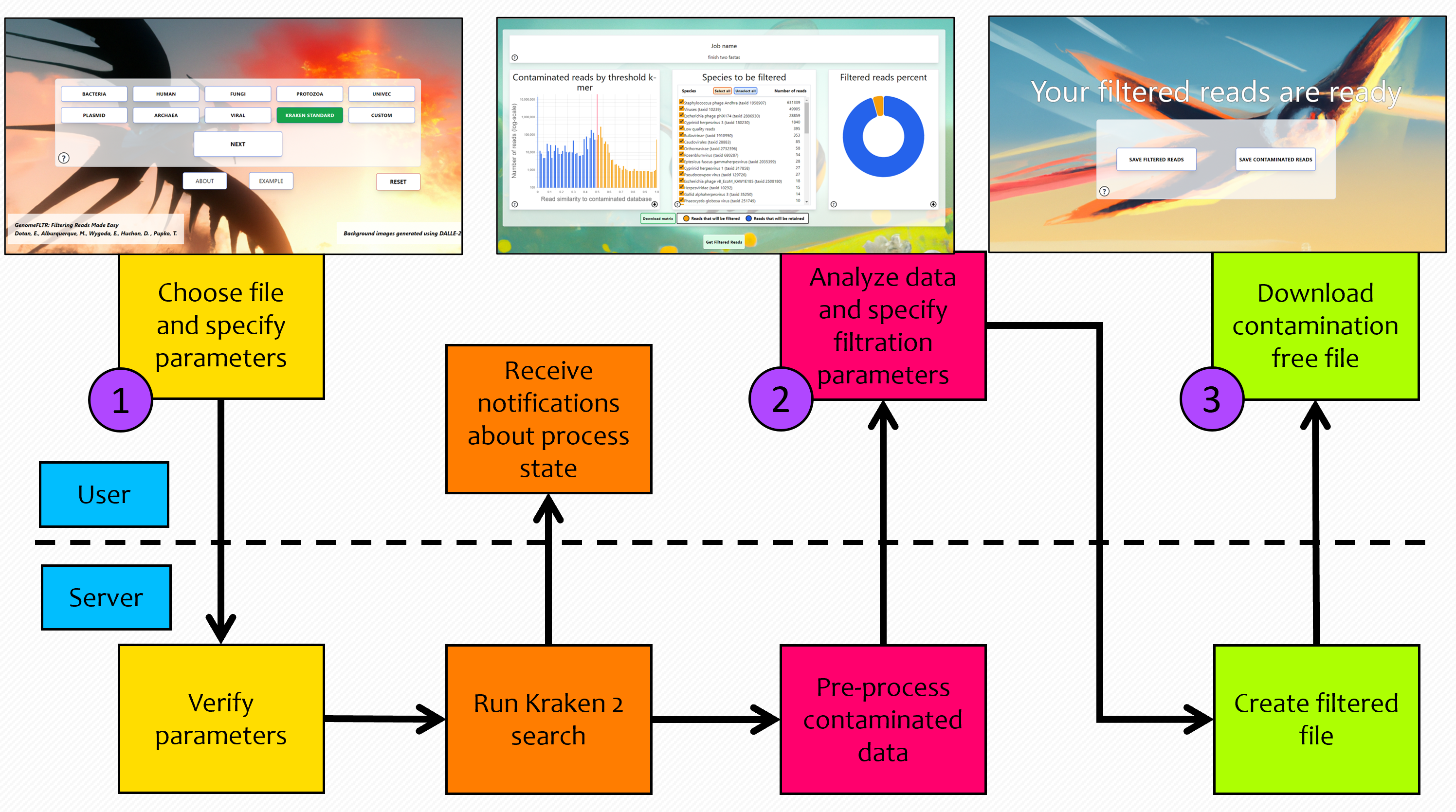
Figure 1: Illustration of the three steps.
Each read is first split into k-mers (k-mers are substrings of the read with length k; for example, 3-mer for the read: “ATGG” will be: “ATG” and “TGG”). To maximize both speed and accuracy, we use the Kraken 2 search engine (Wood, et al., 2019) to query each k-mer (with k = 35) against the selected database. A phylogenetic tree representing the evolutionary relationships within the taxon included in each Kraken database is used to classify hits to either species or ancestral nodes. If a k-mer only matches a single species, it will be assigned to it. If a k-mer matches multiple species, it will be assigned to the most recent common ancestral node of all these species. Note that different k-mers within the same read might be assigned to different nodes of the phylogenetic tree. The output of this step is a file containing, for each of the reads, a list of species or ancestral nodes and the number of k-mers matched to each node.
The output of the previous step is further processed in order to classify each read to a specific node in the tree. To this end, for each read and for each node we define a read-node score, which is the percentage of k-mers mapped to this node divided by the total number of k-mers possible for that read (l – k + 1, where l is the read length). For each read, we identify the node that maximizes the read-node score and assign the read to this node. A tabular description of the number of contaminated reads from each node is provided as interactive visual output by GenomeFLTR as well as a pie chart indicating the percentage of contaminated reads. We also define a read-contamination score, which is the sum, over all nodes of the tree, of the read-node score. This score quantifies the percent of k-mers that were mapped to the contaminated database out of the l – k + 1 total k-mers. The higher the read-contamination score, the more likely it is that the read is a contamination and hence should be filtered.
Another feature implemented in the web server is the filtering of paired-end reads. Each end is first processed independently as described above. Next, the node-score of the pair-end read is the maximum over the two ends. For example, if one end has a read-node score of 0.2 for species X, and the other end the read-node score is 0.75 to species Y, the result of the paired read is a read-node score of 0.75 to species Y. Based on the read scores, the paired reads are either filtered or not, thus, if one end within a pair is considered to be a contamination, the entire paired-end read is discarded.
Output
A histogram illustrating the distribution of the read-contamination score is given as an interactive graphical output by GenomeFLTR. The user specifies a threshold cutoff that determines which reads will be labeled as contamination and which will be retained in the “clean” data. By default, this threshold is set to 0.5. This threshold can be set interactively by clicking on the bars of the histogram. Reads with a score lower than the threshold (this threshold is marked by a red line in the graphical plot) are colored blue and will be retained, while reads colored orange will be filtered once the user presses the “Get filtered results” button.
It is possible that a user chooses to retain reads of specific species. For example, if a user sequenced a metagenomic sample containing multiple bacteria species, and would like to retain only a subset of those bacteria, e.g., bacteria that are known to exist in a specific niche. He can do so, by choosing specific species to retain / filter from the interactive tabular section of the GUI. The pie chart and the histogram are updated accordingly in real-time. We note that in this case, some blue reads (retained reads) could appear to the right of the red bar, which indicates the read-contamination score threshold.
Pressing the “Get filtered results” button initiates the post process, which iterates over the reads and identifies the “cleaned” from the contaminated ones. When the post process is finished, a link to download a compressed file (i.e., a “.gz” file) containing all the non-contaminated reads is provided on the screen and via email to the user.
Example
A video demonstrating the web server usage.
example pageContact
In case of problems or questions, email us at: edodotan@mail.tau.ac.il.